“LNG analysis” typically refers to liquefied natural gas analysis. It involves studying the quality, quantity, and composition of liquefied natural gas. This analysis helps in understanding the properties and characteristics of LNG, which is essential for its storage, transportation, and use.
- Type of gas chromatograph
- Calibration & verification
- Verification
- Calibration and verification gas quality
- Environment for a gas chromatographic system
- Analysis of regasified LNG and retained samples
- Uncertainty of gas analysis
- Raman spectroscopy
- Impurities
- General
- Specifications and measurement of trace impurities in LNG
The analysis of regasified LNG and retained samples involves examining the properties, composition, and quality of LNG after it has been regasified and any samples that have been kept for analysis. This analysis is important for ensuring the safety, quality, and compliance of the LNG before it is used or transported further.
Sampled regasified LNG is analyzed by gas chromatography in order to determine its composition. The reason for this is to be able to calculate the physical properties needed to calculate the energy content. A direct energy content measurement by e. g., calorimeter would be less precise and would also not give the useful compositional information to calculate other properties such as density or Wobbe index.
The arithmetic average of the on-line GC analyses or the average composition of the gas chromatographic analyses of the offline retained samples shall determine the molar composition of the LNG. For the purpose of determining the molar composition, all hydrocarbon components heavier than pentane can be included in the normal pentanes fraction (C+5). Alternatively, components heavier than normal hexanes can be included in the normal hexanes fraction (C+6). The not-yet-normalized (raw) total must be between 99 and 101 mol %.
Also, gas chromatography can be used to determine some impurities like sulphur components at low (ppm) levels. A different set up is often required than for the main components (see below).
Other trace impurities, like mercury, require a different analytical technique. With most impurities sampling is critical and special precautions and sampling materials are required (see below).
All classical techniques used to determine the composition of gas mixtures can be directly applied in the case of regasified LNG.
Many methods exist in the open literature that help technicians to analyze regasified LNG, for example in international standards series, like ISO (e. g., ISO 6974), national institutes like the Energy Institute (IP 337) or methods from institutes like ASTM (ASTM D 1945) or GPA (e. g., GPA 2261).
Note: it is important that, the LNG sample to be analyzed has been vaporized and conditioned correctly to be sure that the sample is truly representative of the LNG (un)loaded and thus the result of the analysis is as well (see Selection principles LNG samples and temperature on tankers“LNG Sampling principles”).
Note (for information only): New developments Alternatives to gas chromatography have been developed with a measurement in gaseous products. A tool measuring speed of sound, thermal conductivity, pressure and temperature can estimate the Calorific Value, Wobbe Index, Density, Compressibility Factor, and other properties. The unit should not be used for compositional analysis but infers a simplified gas composition.
For the direct measurement of LNG density, a «density and speed-of-sound» measurement device has been developed.
Type of gas chromatograph
Among the various arrangements that can be found, the following are given as examples:
- a chromatograph with 2 or 3 columns to separate selectively the components, for instance: one column for N2, C1 to C5 and one column for C6+; or one column for N2, C1, C2 and CO2 and one column for C3, C4 and C5. These arrangements were developed in the 1980’s and are described in the ISO, ASTM, GPA and IP methods;
- alternatively, any modern chromatographic equipment that meets the precision statements for all components to be measured in the ISO, ASTM, GPA or IP methods. A typical refinery gas analyzer will fulfil these requirements.
Calibration & verification
The application of gas chromatography in natural gas is a technique that requires calibration. Calibration aims to establish the relationship between certified values and instrument response (peak area).
There are two possibilities:
- Type 1 analysis in ISO 6974: first determining response functions by means of a multi-point calibration using several calibration standards, followed by regression analysis. These response functions are then used to calculate component mole fractions. Type 1 analyses do not involve non-linearity errors.
- Type 2 analysis in ISO 6974: assumes a linear response function, and subsequent sample analysis is carried out against routine calibrations using a single calibration standard. Because the assumed response function can differ from the true one, type 2 analyses can have non-linearity errors, which shall be evaluated by means of a multi-point performance evaluation carried out in accordance with ISO 10723.
The analyzer system must be calibrated on a regular basis as well as after repair, after maintenance, if verification fails or per vendor recommendation.
Verification
Instrument verification aims to ensure that performance is within predefined limits. The benefit of verification in addition to calibration is that a record of long-term performance of the instrument is kept.
Instead of calibration before and after (un)loading, analyzer verification can be performed.
If the instrument fails to meet the verification criteria a root-cause investigation should be performed. One of the possible remedies could be a recalibration of the instrument.
Calibration and verification gas quality
Calibration shall be carried out with a certified reference gas mixture (CRM, level 2 according to ISO 14111) or better. It should be characterized by a valid metrological procedure for the specified properties, accompanied with a certificate that includes the concentrations of all components and their uncertainties, an expiration date and a statement of metrological traceability. The calibration gas composition must be a close match to the expected LNG quality, or in case of multilevel calibration, cover the expected range of LNG qualities for all components.
Verification should be done with an independent gas mixture which can be of a lower quality than the calibration mixture. A Working Standard Gas Mixture (WRM) (ISO 14111 level 3), in which all relevant components are certified and that contains no «balance gas».
Environment for a gas chromatographic system
The practical requirements for the installation of such a system are the same as those required for any high-accuracy analysis device and mainly involve:
- installation in a closed and temperate (not necessarily air-conditioned) housing, sheltered from the sun, heating sources or draughts, appropriate and constant temperature of calibration gases and sample (injected mass/constant volume);
- permanent and secured electrical supply, without interferences;
- shrouding and earth connections of the electrical connections between the chromatograph and the data system.
Analysis of regasified LNG and retained samples
During a normal Features of cargo delivery LNG/LPG carriersLNG transfer operation (for instance, transfer duration of 12 hours, sampling period duration of 8 hours), the following analysis procedures can be carried out:
- in the case of direct connection between vaporizer and chromatograph: the analyses can be made successively during the whole sampling period, with a frequency equal to the duration of each analysis by the chromatographic system. Example: with one analysis every 20 minutes, 24 analyses are available during the sampling period. For new generation chromatographs, the duration of each analysis is reduced to 5 minutes resulting in 96 analyses;
- in case of periodic filling of sampling containers: one or more (often two) analyses can be carried out successively on each gas sampling container, with a comparison of results, and possible additional analysis or new filling of sampling containers for important threshold component concentrations. This is followed by calculation of the arithmetic average of the percentages of the components determined by the analyses considered for the determination of the average composition of the sample. Example: two analyses on each container filled every hour and calculation of the average, so 8 average analyses during the sampling period;
- in the case of filling of a gas holder (ISO 8943): at the end of the sampling period, three containers are filled, one for each of the seller and buyer and one kept for further investigations (e. g., an independent third party, in case of a dispute). One or more (generally two) analyses can then be carried out on the same sample and retained, if there is no significant threshold component concentration.
In any case, a data set is obtained from the regasified LNG analysis. These data have to be subjected to a statistical process to eliminate outliers, bad analyses, etc. With the remaining data an averaging and normalization treatment is normally done to get the best composition (truly representative) of the LNG volume (un)loaded (see below).
From that composition the Understanding LNG Tank Atmosphere and Material Properties: Key Principles for Safety and Efficiencymain LNG properties may be derived.
Uncertainty of gas analysis
The uncertainty of the gas analysis should be calculated according to ISO 6974-2. Details on the uncertainty calculation can be found in Recommended methods for calculating LNG“Uncertainty of the energy transfer determination”.
Raman spectroscopy
At the time of publication of this sixth edition, a different metrology device for analysing LNG composition with direct analysis in the LNG transfer line(s) is undergoing some pilot testing.
Raman spectroscopy (see below) is an analytical technique that uses monochromatic light to excite and identify the vibrational modes of molecules. Each mode of each molecule generates a shift in the frequency of the scattered light. By analyzing the frequency and intensity of the scattered light, the sample’s composition may be determined. The scattering interaction is so short-lived that the measurement is independent of the flow rate of the sample. The technique is viable for all phases of matter and may be effectively used on mixed phase samples. Since the intensity of scattered light is dependent on number of molecules that participate, the best results are achieved with solids, liquids and high pressure gases.
DIRECT IN-LINE ANALYSIS WITH RAMAN SPECTROSCOPY
The technology
Raman spectroscopy is a form of vibration spectroscopy where a material of interest is illuminated by a highly monochromatic light source, typically a laser, and the resulting scattered light is analyzed. When light interacts with a molecule, most photons are elastically scattered. The scattered photons have the same energy (frequency) and, therefore wavelength (colour) as the incident photons. However, a small fraction of light (approximately 1 in 108 photons) is scattered at optical frequencies different from, and usually lower than, the frequency of the incident photons. The process leading to this inelastic scatter is termed the Raman effect, discovered in 1928 by Indian physicist Chandrasekhara Venkata Raman who was awarded the Nobel Prize for physics in 1930.
The energy of a molecular vibration mode depends on the molecular structure of the material. A Raman spectrum is, in effect, a «molecular fingerprint» unique to that compound (see Figure below).
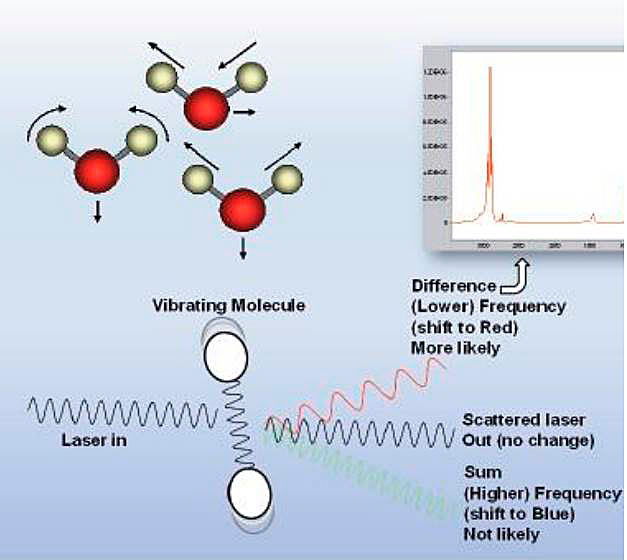
The spectrum can be analyzed to obtain information on the molecular components of the material being illuminated as well as their relative abundance. The signal strength is strongly dependent on the density of the sample, so it is advantageous to measure the liquid directly. This also avoids the need for the phase change or molecular separation common with other analytical methods. Due to the fact that the Raman effect is a relatively weak process, with the currently available technology it is typically not suitable for trace (low ppm) analysis of higher (C7+) components in LNG.
The adaptation to LNG
For LNG analysis a cryogenic optical probe and an internal calibration process were developed. The latter allows the analyser to perform diagnostics automatically.
Via a fibre optic cable and the optical probe, the laser light is introduced into the LNG pipeline through a sapphire window at the end of the probe. The actual molecular excitation takes place approximately 300 to 400 microns off the window. As there is no «wet chemistry» taking place there is no need for cleaning the device or other regular maintenance issues. The probe contains a series of optical components for filtering the light and is connected to a base unit by a length of fibre optic cable.
The fibre optic cable is terminated in the analyser base unit which can be installed in the field or inside a technical building, e. g. a process control room. An initial calibration of the equipment is performed at the factory prior to shipment. Once installed on site, a simple calibration of the fibre transmission intensity is all the calibration that is needed. Analyser maintenance typically occurs on an annual basis.
Laboratory and LNG field testing allowed for the development of a mathematical model to convert the Raman spectrum to a composition and energy content. This model will be enhanced as additional field data is gathered, thus eliminating the variation that results from individual calibrations of current composition analysis systems that are required by gas chromatographs.
Field testing
Data was collected on several LNG carrier unloading operations. The results of the measurements indicated that 2-sigma repeatability is better than 0,1 BTU and accuracy is better than 0,5 BTU when compared to gas chromatographic data taken at the same time (see Figure below).
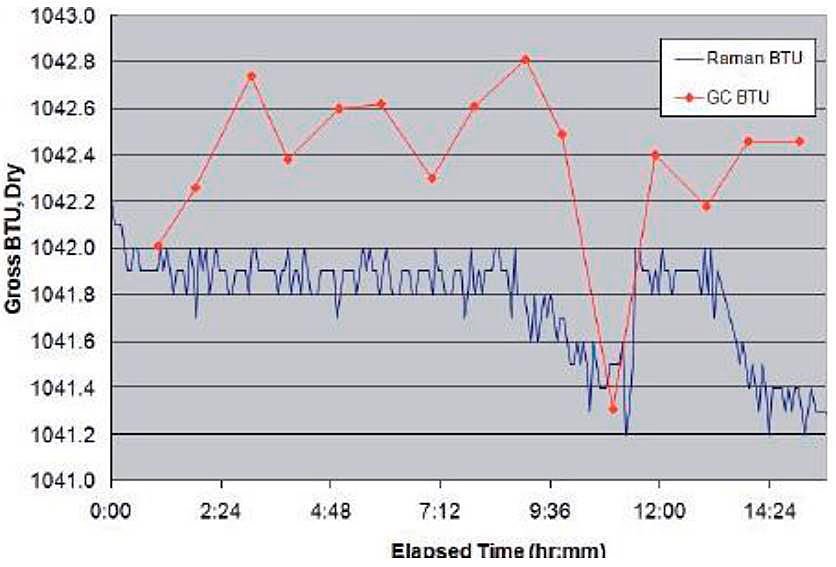
After analysis, the bulk of the 0,5 BTU offset appears to be due to the BTU value assigned by the chromatograph to C6+ components, which are not (yet) incorporated in the current Raman Model.
Future model refinements, which will include the effects of the C6+ components should be able to close the offset. This may be further improved through the use of a more sophisticated model that incorporates more extensive field and laboratory analysis.
For LNG, Raman spectroscopy is accomplished through the use of an optical probe that is inserted into a product stream. The probe is typically of stainless steel construction with a small sapphire window at the tip to allow the incident laser light and scattered light to pass to the analyzer. The probe must be designed to operate at cryogenic temperatures with no loss of function to the enclosed optics. The light is passed to and from the probe through fiber optic cables in a suitably rugged cable assembly. The most basic Raman analyzer consists of a laser, spectrograph and a processor to operate them. The laser must be sufficiently stable to allow the shift in light to be consistently measured and be powerful enough to deliver close to the maximum allowable optical power to the probe tip. The spectrograph must also be capable of measuring the frequency and intensity of light to great precision. Since Raman scattering is a non-contact, non-destructive technique, calibration may be accomplished without need for custom gas or liquid samples. An instrument is calibrated by characterizing the wavelength and intensity of the laser and the sensitivity of the spectrograph. This can be accomplished with stable physical references such as neon gas or diamond crystals.
The potential of Raman scattering as an analytical technique for LNG is in its ability to measure a liquid directly without a phase change to a gas. Raman is not well suited for trace analysis of components such as sulphur.
Pilot field testing on different locations is still ongoing in view of further validation and eventual acceptance in the LNG industry.
Impurities
General
Possible contamination of LNG is a concern because it may have safety and reliability consequences in relation to:
- LNG transfer systems;
- the sampling system and analytical instruments;
- systems and equipment in which the LNG is to be processed;
- systems and equipment exposed to vaporized LNG.
This information given here is just to raise awareness of this issue. It is realised that some contaminants and/or impurities will often be present at some (expected) level and the risk of particular consequences may depend on the level. It should be noted however that some contaminants/impurities may be cumulative.
Corrective and proactive measures may differ from site to site. The source of contamination may be at the location where the LNG is produced, in the transport carrier or possibly in the receiving system in which the LNG is processed and vaporized.
Examples of contaminants and their potential impact include:
- water: when exposed to LNG, water or water vapor turns into a solid (ice) which can block sampling systems, valves and instrument taps as well as damage equipment;
- particulates: metal shavings, welding debris, insulation, sand, wood and cloth are typical examples of particulate material. If inert, the most common problem with particulates would be blockages and damage to equipment;
- sulphur: a sampling point that utilizes copper or copper alloys may be damaged by contamination with sulphur and/or the measurement of trace sulphur compounds may be impaired by their chemical reaction with copper;
- mercury: traces of mercury may damage aluminium components by chemical reaction with the aluminium. A release of gas due to a resultant failure of the aluminium is an example of a possible safety consequence;
- other hydrocarbons: a sampling or piping system that contains, for example, traces of LNG, may result in erroneous analysis, or otherwise in «LNG out of specification». Moreover, due to the limited solubility of butanes and higher paraffins in LNG, too high concentrations of these may also solidify and clog sampling systems. Lubricating oil and seal oil contaminants if present will be in the form of hard solids;
- Inert gases: nitrogen and air may be present in both sampling systems and piping systems from, perhaps, inadequate or poor purging operations. Moreover, the presence of oxygen from the air may present a safety hazard;
- CO2 exposed to LNG, turns into a solid similarly to water (ice) and may block sampling systems and damage equipment. Traditionally, as measured by Davis et al (1962), the solubility of carbon dioxide in the liquid and vapor phase has been 340 ppm and 85 ppm respectively at 111,5 K and 1 bar. Recent theoretical research by Shen & Lin et al (2011-2012) and experimental research by Gao et al (2012) into CO2 solubility in liquid CH4/N2 mixtures at cryogenic temperatures have identified that at any stage of the liquefaction process it is estimated that only slightly more than 100 ppm of carbon dioxide could be present.
In establishing the major constituents of the LNG (for GHV determination), the danger potentially exists to ignore (trace) contaminants and/or impurities because they are normally not present at levels that exceed tolerance. The consequence may be damage to equipment, may lead to safety problems and possibly result in customer rejection of the LNG.
Specifications and measurement of trace impurities in LNG
Trace impurities in regasified LNG which are often specified in LNG contracts are carbon dioxide, sulphur components (hydrogen sulphide, carbonyl sulphide and mercaptans) and mercury. The trace impurities are normally in the range 0-1 mg/m3(n) or, in case of mercury as low as 5 ng/m3(n). Sampling cannot be done in normal cylinders for these trace impurities since they are chemically reactive and will be absorbed by the wall of the sample cylinder.
The determination of trace impurities requires a special approach. This can hardly be underestimated; set-up, operation and maintenance are an area for specialists. All aspects are critical: sampling, calibration and analysis. Validation and verification of results is strongly advised before using the results of the analyses for commercial purposes.
Carbon dioxide. The specification limit for carbon dioxide is often around 0,01 mol % (100 ppm). The carbon dioxide content is normally determined by gas chromatographic (GC) analysis which is capable of analyzing down to this limit or even lower.
Sulphur. Sulphur impurities are normally specified at the 0-25 mg/m3(n) level.
Sulphur can be specified as total sulphur and/or as specific sulphur containing components: hydrogen sulphide (H2S), carbonyl sulphide (COS) and mercaptans (RSH, where R is an alkyl group, e. g., methylmercaptan, CH3SH or ethylmercaptan, C2H5SH).
Sampling for trace sulphur components is not so easy; special precautions are needed in order to avoid adsorption of sulphur components to the wall of the sampling system devices. Sampling in containers is preferably made according to the standard method described in ISO 10715. The interior face of the sample cylinders must be made out of a material which doesn’t react with sulphur components. Even then, the sample cannot be kept for more than 8 days.
The commercially available materials «silicosteel» or «sulfinert coated steel» are suitable for this application but are very expensive.
Total sulphur. Total sulphur can be determined by combustion techniques, where all sulphur is converted into SO2 which is trapped and quantified. Combustion techniques are often specified in LNG contracts and are cost effective but are not the safest to apply. Many accidents have occurred due to the rather violent combustion of gas in an oxygen/hydrogen flame.
Newer instrumental techniques like microcoulometry (ASTM D3120), pyrolyses/chemoluminescence, or hydrogenolysis/rateometric colorimetry (ASTM D4045), GC/chemoluminescence (ASTM D5504) or UV fluorescence (ASTM D6667) are preferred.
Sulphur components. In order to determine the sulphur components separately in a gas, these components must be separated first. A gas chromatograph can be used for this in combination with a sulphur specific detector. The commonly used detectors (TCD – not sensitive enough for trace levels and FID – no signal at all) are not suitable. Several sulphur specific detectors are available and are described in ISO 19739 (Determination of sulphur compound by GC). Equipment for these measurements is expensive and requires specialist skills. On-line measurement is, in principle, also possible.
In principle the total sulphur can also be calculated from the components present, under the assumption that all sulphur components are detected and measured by the GC (which is normally the case, of course).
However, new instrumental techniques as mentioned before for the determination of the total sulphur content such as UV fluorescence (ASTM D6667) are preferred rather than using GC’s.
Mercury. Mercury can be determined by ISO 6978. For low levels of mercury, the procedure is described in part 2: Sampling of mercury by amalgamation on gold/platinum alloy. This method is described in the range 10-100 000 ng/m3(n) (sampling at atmospheric conditions). Mercury is normally in the form of its metal or light organic mercury components (e. g., methyl mercury). These forms are trapped effectively by amalgamation.
Mercury sampling is very critical because of the adsorption properties of mercury. Sampling lines should be as short as possible (see also ISO 10715 for materials, preferably less than a meter in length), preferably heated (80 °C) especially if the gas pressure is high (above some 20 barg) to prevent possible condensation of hydrocarbons and should be flushed for a very long time (preferably even continuously at a low rate, e. g. 2 l/min).
Read also: Selection principles LNG samples and temperature on tankers
Commercial equipment is available on the market for mercury determination but doesn’t fit strictly with the recommendations of ISO 6978 which has been developed mainly for high pressure gas measurements. However, for mercury levels in LNG, commercial equipment is efficient if precautions are taken.
As with sulphur impurities, gaseous sampling for mercury in a cylinder is difficult. It is not reliable for mercury at low levels which will be adsorbed by the cylinder wall in a very short period of time causing erroneous results.